量子化學如何選擇計算方法與相應的基組?
Gaussian是目前應用最廣泛的量子化學計算軟件之一,既可進行半經驗計算,也支持從頭算與密度泛函理論等多種計算類型。該程序幾乎涵蓋了分子體系研究的各個方面,可用于探索分子能量與幾何結構、過渡態的構型及反應勢壘、化學鍵性質與反應能量、分子軌道與電荷密度分布、偶極矩與靜電勢、多極矩特征、振動頻率、紅外與拉曼光譜、核磁共振(NMR)參數、極化率與超極化率等光學性質,以及熱力學數據和反應路徑等。
在實際計算中,選擇合適的計算方法與基組是獲得準確可靠結果的關鍵。不同的方法在計算精度與資源消耗上差異顯著,了解它們的原理和適用范圍有助于根據研究體系與目標做出最優決策。以下將對Gaussian中常用的幾類計算方法進行簡要介紹與選擇建議。
一、計算方法的選擇
半經驗方法(AM1、PM3、CNDO、INDO、MINDO)
通過引入實驗參數簡化計算,速度快但精度有限。
適合:
大體系有機分子的幾何優化;
初步構型篩選與趨勢判斷。
不適用于:
含金屬或強電子相關體系;
高精度能量計算。
?建議:僅在體系龐大、計算資源有限時用于初步優化。
2. 從頭算(Ab initio)方法——Hartree-Fock (HF) 系列
HF方法完全基于量子力學原理,不含經驗參數。
它通過自洽場(SCF)計算電子波函數,是更高精度方法的基礎。
適合:
小體系的幾何結構優化;
作為MP2或CCSD計算的前導步驟。
不足之處在于未考慮電子相關,能量偏高。
?建議:用于結構優化或提供參考波函數。
3. 密度泛函理論(DFT)方法
DFT以電子密度為核心變量,在效率與精度之間取得良好平衡,是目前最常用的計算方法。
常見泛函及特點:
B3LYP:經典穩健,適用于大多數有機分子體系;
M06系列:對金屬、π–π作用、非共價相互作用有較好表現;
wB97XD:考慮長程修正與色散作用,適合大體系或弱相互作用研究。
?建議:B3LYP作為默認起點,若體系含金屬或弱相互作用明顯,可選M06或wB97XD。
4. MPn微擾理論(MP2、MP3、MP4…)
在HF基礎上逐步引入電子相關修正:
MP2:計算精度高且成本適中,是最常用的相關修正方法;
MP3/MP4:理論上更精確,但計算量急劇增加,實際應用較少。
?建議:MP2適用于中小體系的高精度能量計算。
5. 耦合簇(Coupled Cluster, CC)方法
被譽為“量子化學的金標準”,計算精度極高。
CCD:僅考慮雙激發;
CCSD:加入單激發修正;
CCSD(T):進一步微擾三激發,是目前最精確的主流方法之一。
計算量非常大,通常僅用于小分子的基準計算。
?建議:當需與實驗嚴密對比或驗證其它方法可靠性時使用CCSD(T)。
二、基組的選擇
最小基組(Minimal Basis Set)
代表:STO-3G
每個原子軌道只用一個高斯函數近似;
計算量極低,但精度有限。
?用途:快速測試或教學演示,不推薦科研正式計算。
2. 分裂價基組(Split-Valence Basis Set)
代表:3-21G、6-31G、6-311G
將價電子軌道拆分為多個函數,以更好描述電子分布;
是結構優化與能量計算的常用起點。
?建議:6-31G或6-31G(d) 是入門常用基組,可平衡精度與速度。
3. 極化函數與彌散函數
極化函數(d, p, f):用于更精確描述化學鍵方向性與分子畸變(如彎曲鍵、過渡態);
彌散函數(+或++):用于描述電子云分布較寬的體系(如陰離子、氫鍵、π–π作用)。
?示例:
6-31G(d,p):常規優化首選;
6-31+G(d,p):適合含陰離子或弱相互作用體系。
4. 相關一致基組(Correlation-Consistent Basis Set)
代表:cc-pVDZ、cc-pVTZ、cc-pVQZ
由Dunning提出,能系統改善電子相關描述;
常用于高精度計算(MP2、CCSD等)。
?建議:MP2或CCSD計算中使用cc-pVTZ可獲得較可靠能量。
5. 有效芯勢(ECP)基組
代表:LANL2DZ、SDD、Def2系列
通過勢函數近似內層電子效應,顯著降低含金屬體系的計算量。
?建議:
含過渡金屬體系 → 使用LANL2DZ或Def2-TZVP;
想兼顧精度與速度 → Def2-SVP是不錯選擇。
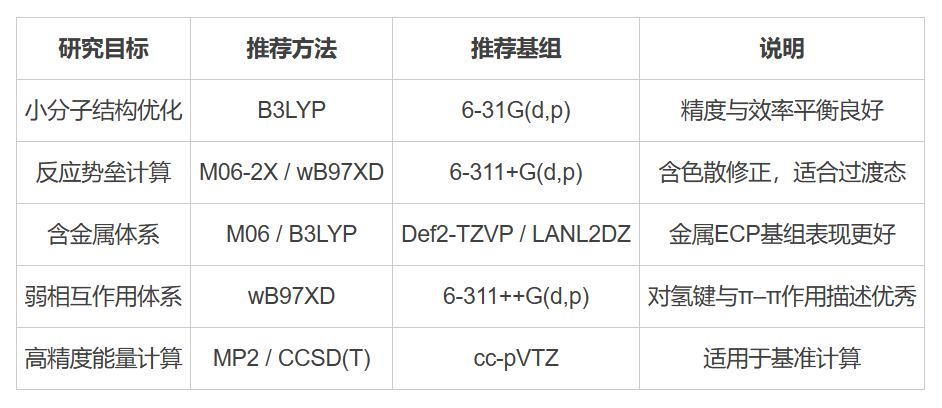
三、常見方法與基組搭配建議




 投訴建議
投訴建議
提交